圆锥交叉点(MECI)的优化
MECI点是光化学反应中关键的结构,然而S0与S1的CI点由于存在绝热近似失效的问题,难以使用TDDFT来描述,常常用多参考方法来计算。后者虽然可以避免参考态波函数不稳定的问题,计算量却比DFT大的多,适用于小体系。若遇到大体系,还有一些方法是可以考虑的:
- 纯泛函:对于能量裂分比较大的情况,低HF成分的泛函可以凑合用。
- SF-TDDFT:通过以三重态作参考态避免参套态波函数不稳定的问题,缺点是会引入自旋污染。GAMESS、ORCA支持。
- MRSF-TDDFT、SA-SF-TDDFT:是前者的升级版,可以解决自旋污染的问题,但MRSF-TDDFT只有GAMESS支持,SA-SF-TDDFT只有Q-Chem支持。
纯泛函(sobMECP)
从原理上讲,DFT没有办法正确描述圆锥交叉点,只能勉强描述圆锥避免交叉。要使用sobMECP搜索圆锥避免交叉,首先要把能量收敛判据给删掉。找到:
1
2
3
4
5
IF (PConv(1) .and. PConv(2) .and. PConv(3) .and. PConv(4) .and. PConv(5)) THEN
Conv = 1
ELSE
Nstep = Nstep + 1
END IF
改为
1
2
3
4
5
IF (PConv(1) .and. PConv(2) .and. PConv(3) .and. PConv(4)) THEN
Conv = 1
ELSE
Nstep = Nstep + 1
END IF
然后准备头文件。对于静态相关比较强的圆锥避免交叉,最好用纯泛函来描述:
1
2
3
4
5
6
7
8
%mem=192GB
%nproc=64
%chk=S1.chk
#n MN15L/6-31g* td force guess(read)
Second State
1 1
由于使用TDDFT计算state2,需要一个extract_energy2文件:
1
2
3
4
5
{
if ($3 == "E(TD-HF/TD-DFT)") {
print $5
}
}
完成后,依次运行:
1
2
3
./prepare.sh
./runfirst.sh
./runMECP.sh
这个不太实用,因为CI点附近势必出现前线轨道简并的情况,而这又将引起Gaussian的L801报错:
1
2
3
**** Fatal Problem: The smallest alpha delta epsilon is 0.57953183D-03
Error termination via Lnk1e in /work/home/gaus12/apprepo/gaussian/16-hy/scripts/../app/g16/l801.exe at Tue Feb 25 10:39:30 2025.
采用IOp(8/11=1)可以避免出现该报错时终止任务,但轨道能量差在计算时需要承担分母,接近0会产生导数数值不稳定的问题,很可能最终无法收敛。
SF-TDDFT
参考态需要三重态。准备好xyz文件,使用ORCA输入文件:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
! CI-opt WB97X-D3 def2-SVP RIJCOSX def2-SVP/C miniprint
%maxcore 3000
%pal
nproc 32
end
%tddft
sf true
nroots 5
iroot 2
jroot 1
end
%geom
maxstep 0.05
trust -0.05
end
*xyzfile 0 3 probe.xyz
NOTE:SF-TDDFT的基态是1号,第一激发态是2号。
该方法不是纯态方法,存在自旋污染问题,时常难以分辨哪个是单重态,只能凑合用:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
---------------------
SF-TDA EXCITED STATES
---------------------
the weight of the individual excitations are printed if larger than 1.0e-02
(SPIN-FLIP GROUND STATE)
STATE 1: E= 0.027946 au 0.760 eV 6133.4 cm**-1 <S**2> = 0.147846
96a -> 100b : 0.014731 (c= -0.12137004)
99a -> 100b : 0.044605 (c= 0.21119915)
101a -> 100b : 0.872210 (c= 0.93392172)
101a -> 102b : 0.036161 (c= -0.19016046)
101a -> 104b : 0.014738 (c= 0.12139931)
STATE 2: E= 0.027972 au 0.761 eV 6139.2 cm**-1 <S**2> = 1.169462
99a -> 101b : 0.026155 (c= 0.16172449)
101a -> 101b : 0.921675 (c= 0.96003905)
101a -> 106b : 0.011819 (c= 0.10871489)
可以看到STATE2的自旋污染很大,<S**2>达到了1.17,指认单重态或是三重态都感觉不太合适。
MRSF-TDDFT
GAMESS的收敛性不太好,参考态波函数最好由Gaussian准备。首先,要在RODFT下计算一个三重态作为参考态。
1
2
3
4
5
6
7
8
9
10
11
12
%chk=SRR.chk
%mem=96GB
%nprocshared=32
#p ROBHandHLYP/6-31g* nosymm int=nobasistransform
title
1 3
C -0.59562400 -0.17774100 0.00000000
C 0.04669800 -0.09509800 1.24125300
...
这里用的基组一会还要做opt,不要太大
将收敛的波函数传给GAMESS。这一步要用到MOKIT,需要提前装好。
1
fch2inp name.fchk -mrsf
产生的inp文件中,MWords是每个核分配的内存,类似于ORCA的%maxcore,但单位为MW(1MW=8MB),可能需要修改:
1
2
3
4
5
6
7
8
9
10
11
$CONTRL SCFTYP=ROHF RUNTYP=ENERGY ICHARG=1 MULT=3 NOSYM=1 ICUT=11
MAXIT=200 DFTTYP=BHHLYP TDDFT=MRSF $END
$SYSTEM MWORDS=600 $END
$SCF DIRSCF=.T. $END
$GUESS GUESS=MOREAD NORB=640 $END
$DFT NRAD0=99 NLEB0=590 NRAD=99 NLEB=590 $END
$TDDFT NSTATE=5 $END
$DATA
GAMESS inp file produced by MOKIT,na=136,nb=134,nif=640,nbf=640
...
运行命令:
1
rungms SRR-CI.inp 00 64 > SRR-CI.gms
其中第一个数字是版本号,第二个数字是并行核数。该文件是单点计算,可能耗时稍高于普通TDDFT单点。由于使用了Gaussian传递轨道,SCF通常会在几圈内收敛。然而,由于GAMESS的SCF收敛性并不出色,也可能出现这种情况:
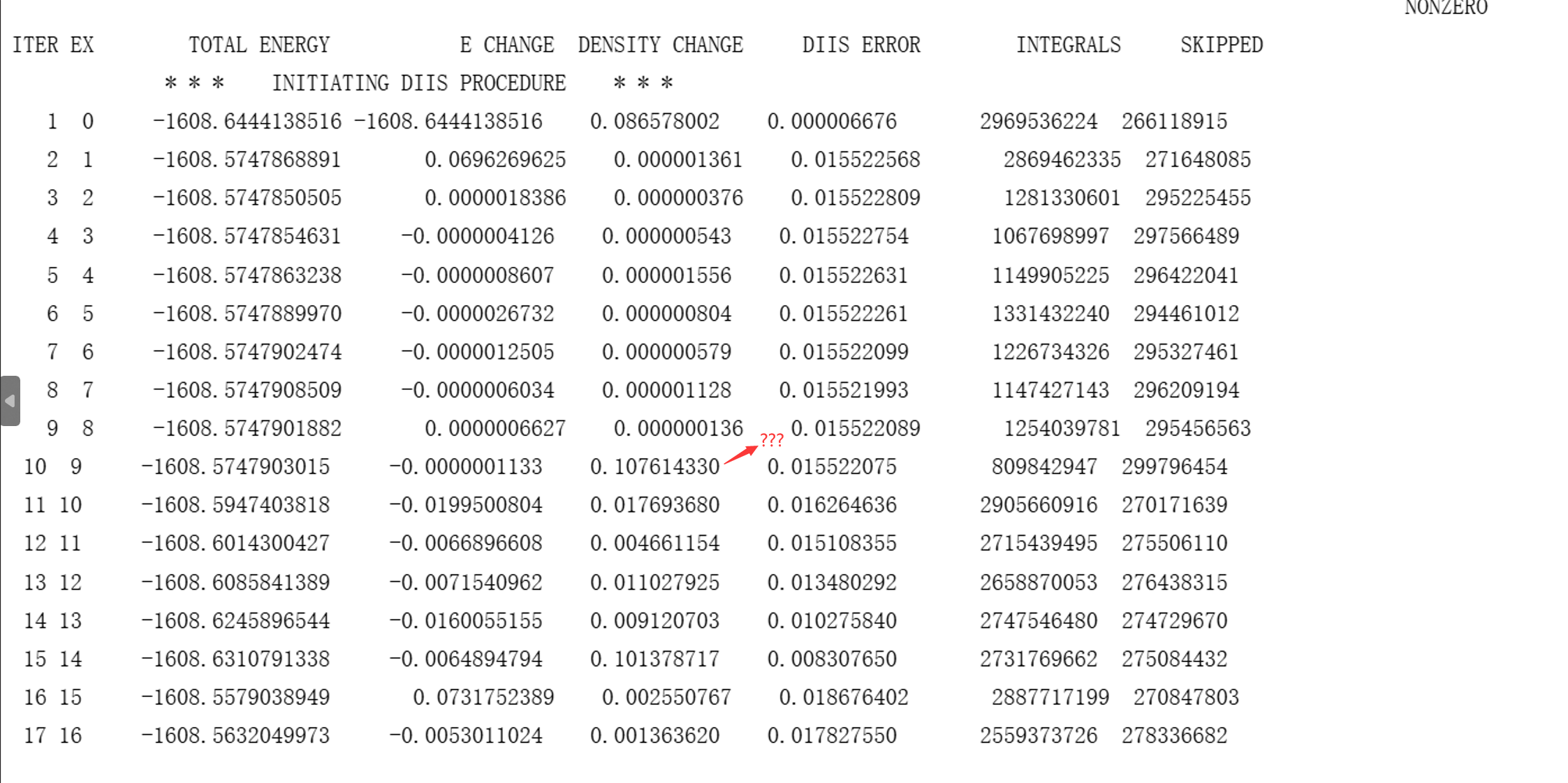
笔者将该GAMESS计算45分钟迭代200圈后收敛失败的结构交给Gaussian,被Gaussian用1分45秒走了30圈拿下,只能说Gaussian不愧是主流量化程序。这种情况下,也许可以尝试切换SCF算法:
1
$SCF DIRSCF=.T. DIIS=.F. SOSCF=.T. $END
如果还是不行,建议求助大师。这里假设顺利运行完毕,在输出文件中找到:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
----------------------------
SUMMARY OF MRSF-DFT RESULTS
----------------------------
STATE ENERGY EXCITATION <S^2> TRANSITION DIPOLE, A.U. OSCILLATOR
HARTREE EV X Y Z STRENGTH
1 NEGATIVE ROOT(S) FOUND.
1 A -1608.7798225490 -1.153 0.0000 0.0000 0.0000 0.0000 0.0000
0 A -1608.7374375718 0.000 (REFERENCE STATE)
2 A -1608.6970167917 1.100 0.0000 0.0000 -0.0000 6.4967 2.3300
3 A -1608.6757530133 1.679 0.0000 -0.1573 -0.0340 0.0000 0.0018
4 A -1608.6528024556 2.303 0.0000 -0.0000 -0.0000 -0.9923 0.0834
5 A -1608.6417893495 2.603 0.0000 -1.0486 -0.1358 0.0000 0.1029
要优化S0与S1的势能面交叉点,即搜索态1和态2的圆锥交叉,更改输入文件,将RUNTYP设为CONICAL,并添加$CONICL字段:
1
2
3
4
5
6
7
8
9
10
11
12
$CONTRL SCFTYP=ROHF RUNTYP=CONICAL ICHARG=1 MULT=3 NOSYM=1 ICUT=11
MAXIT=200 DFTTYP=BHHLYP TDDFT=MRSF $END
$SYSTEM MWORDS=600 $END
$SCF DIRSCF=.T. $END
$GUESS GUESS=MOREAD NORB=640 $END
$DFT NRAD0=99 NLEB0=590 NRAD=99 NLEB=590 $END
$CONICL OPTTYP=PENALTY IXROOT(1)=1,2 SIGMA=8.0 $END
$TDDFT NSTATE=5 $END
$DATA
GAMESS inp file produced by MOKIT,na=136,nb=134,nif=640,nbf=640
...
- OPTTYP设置的是优化算法,PENALTY是对CI搜索比较(相对)好用的惩罚约束算法。
- SIGMA是惩罚项的拉格朗日乘数,默认为3.5
- IXROOT(1)设置的是要搜索的根。
设置好后,再次提交计算。这玩意很难收敛,也不知道是GAMESS的问题还是圆锥交叉本身难找的问题。可能的解决办法:
- 如果你期望的结构是圆锥避免交叉,去临时文件目录找到输入文件同名的.dat文件,其中有当前结构坐标。把该坐标复制出来,写成RODFT输入文件,重复上述步骤,并调小SIGMA值再提交再次提交计算。
- 如果你期望的结构是圆锥交叉,调大SIGMA值再次提交计算。
- 使用OPTTYP=BPUPD来切换为默认几何算法。注意,此时SIGMA设置无效!